-
2016���Ї��tˎ�ĸ������������tˎ�ИIͶ�Yǰ���A�y
2016/5/25 11:06:09����Դ:�Ї��a�I�lչ�о��W�������w���� �� С�����ղر��������ӡ�����P�]��
������ʾ�����˽�Q�҇�ˎƷ�I��O�ܲ�����������o�ȽY����ì�ܵ��T�����}����2013 �����҇�����Ժ������ʳƷˎƷ�O���������ѽ�������һϵ�иĸ�����g�����P���T��������14 �l���ߣ��ĸ�Ҳ����“����——ԇ�c&md�������˽�Q�҇�ˎƷ�I��O�ܲ�����������o�ȽY����ì�ܵ��T�����}����2013 �����҇�����Ժ������ʳƷˎƷ�O���������ѽ�������һϵ�иĸ�����g�����P���T��������14 �l���ߣ��ĸ�Ҳ����“����——ԇ�c——�ƏV”����������M��Ŀǰ��������������ˮ�^���҂��Д࣬2016 �ꌢ�ɞ�����ԁ����m�ĸĸ������_ʼҊЧ���P�I�r�ڣ����H��������ˎ�|����������@��Ӱ푣������tˎ��I������Ͱlչ��ˎƷ�ИIδ���lչ��֎����ش�׃�
����һ��������GMP �˜ʣ���ˎ��Ӳ���OʩҪ���@�����
����1����GMP �˜��Mһ�������ˎ�����a�Oʩ���T��
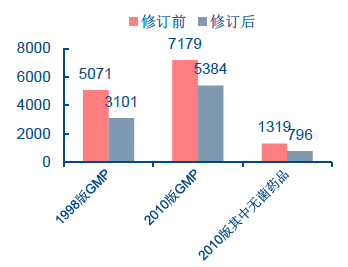
����Ŀǰ�҇�ˎƷע�Ժ����a�h������Ҫ�ıO���ֶ���GMP �J�C�ƶȡ�����ȫ�Q�ǡ�ˎƷ���a�|������Ҏ������Good Manufacture Practice������ˎƷ���a�ĸ����h������ԭ�ϲ�ُ��ˎƷ���a���|���z��Լ�ˎƷ�N�۵��^�̾��бO�ܣ��LJ��Hͨ�õ�ˎƷ���a�|������˜ʡ�1988 ���ԁ��҇��l�����_ʼ����GMP Ҏ���J�C��1998 ���M���˵�һ���������ь�ˎƷ��I��GMP �J�C�˜��m���M������ߡ�1998 ��GMP ��ӆ���J�C��2004 ��6 �µY��������3101 ����Iͨ�^�J�C����2011 ���ֹ����1998 ����ӆˎƷGMP �J�C��I���� 19107�lӛ䛣��@һ�A��GMP �J�C�ī@�����ѽ��^�ߡ�
������ˣ����Mһ�����M�҇�ˎƷ�a�I�����lչ��2011 ��ʳˎ�O������һ���о���ӆGMP�˜ʣ������������l���M�����W�˵ȇ��H���M�˜��~�M����ˎƷ���a��Ҫ���M���^����ȵ�����������ʳˎ�O���֡��P��؞�،�ʩ<ˎƷ���a�|������Ҏ��>��֪ͨ��Ҏ�����҇���ȫ��ˎƷ���փ����քe�M����GMP �J�C����һ���o��ˎƷ���a��I����2013 ��12 ��31 ��ǰ�_������ӆˎƷGMP Ҫ�ڶ�������ȫ��ˎƷ���a��I����2015 ��12 ��31 ���_������ӆˎƷGMP Ҫ��
GMP �˜���ӆǰ����I����
������GMP �ԁ����J�C��r
����2����GMP �˜ʵ��ƏV�����´�����Сˎ�����˳�
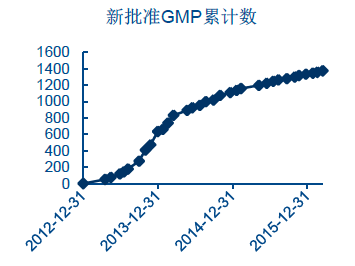
������GMP �o��I�A��������r�g�����ѽY�����AӋ���m���мs30%��ˎ�����˳����Ěvʷ����������D10 ��ʾ���҇���һ�Ό�ʩ1998 ���GMP �J�C��25%��ˎ��ֱ���˳����Ј�����GMP ��ʩ��һ���o��ˎƷ���a��I����Ҳ���F���@���»�������2013 ��Ĕ������҇��o��ˎƷ���a��I��Ӌ1319 �ң����������GMP �J�C��Ո��ֻ��870 �ң�ռ��66%����Kͨ�^�z���ֻ��796 �ң�ռ��60.3%�������ڸ���Ҏģ������ȫ��ˎƷ���a��I��ʳˎ�O�������ƶ��C�l�r�A��������r�g���ڣ��ƶ���2011 ���C�l����2015 ��12 ��31 �ղ��Ǚz����K���ޡ�
�������ϣ����y�㣬��GMP ��������a�������M���^�ߣ�����һ�l���a����ƽ��֧���s��500 �f������10%ƽ���������㣬���ڮaֵ��5000 �f�����µ�ˎ����f������һ�l���a�����M�þͳ��^����Iһ���ӯ��������ʳˎ�O���������Ĕ������Թ�����Сˎ��ƽ���������a������3 �l����ˣ�������Сˎ�������R“�ǻ��M������������칤�S���_�ˣ�߀���P�Tͣ�a”�ĕr����x����ߡ����ϣ��ܶ���Сˎ�����^ȥ�����������������������a���a���棬�@����2015 ��״��ށ��R֮�������в��֎��������N�ۡ���ˣ����M��2016 ���_ʼ���������M����GMP �˜ʵ���Сˎ���_ʼ�M��ͣ�aȥ����A�Σ�һ���@���^�̽Y������I�����R�Pͣ����۵ľ��档
��������ᘌ���ˎ�_��ʷ������R�������˲飬Ӱ���Iδ���F�����A��
����ᘌ���ˎƷ�N��ʳˎ�O�����_����һ݆ʷ���������R�������˲顣2015 ��8 ����Ժ�l�����P�ڸĸ�ˎƷ�t����е���u�����ƶȵ���Ҋ�������؏��{��Ҫ����҇�ˎƷ�����˜ʡ��͡���Ҋ����sͬ�r�ڣ�����ʳˎ�O���ְl���ˡ��P���_չˎ���R��ԇ���Բ�˲鹤���Ĺ��桷�������桷���{�����Դ�^�ϱ���ˎƷ��ȫ����Ч������ʳƷˎƷ�O���������֛Q��������������a���M�ڵĴ���ˎƷע����Ո�_չˎ���R��ԇ���˲飬��������Q�҇�һ����“��ˎ”�R���|�����ߡ�����������١�ע����Ո�e����һϵ�І��}��
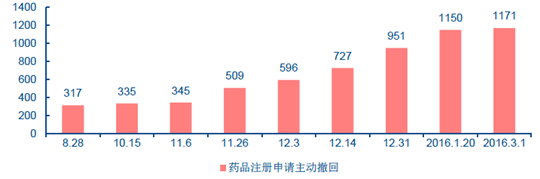
����“ʷ�����”�˲錧�´������ע����Ո���أ�ֱ��Ӱ���I��δ���F�����A�ڡ���݆�˲�ć���̶ȳ��^�˴�ǰ�Ј����A�ڡ�2015 ��ף�CFDA �����˃�݆���Č���Y�������ڳ�z��11 ����I��24 ��ˎƷע����Ո���R��ԇ��ȫ���І��}�������������Pע�ԡ�����е�100%�ܽ^�ʇ����������ИI���^�������I��m��������ˎע����Ո�����P���_չˎ���R��ԇ���Բ�˲鹤���Ĺ��桷�l���r�����ע����Ո�Ĺ���1622 ��ˎƷƷ�N��������CFDA �Wվ���濂�Y����ֹĿǰ1622 ��Ʒ�N�У���ȥ193 ������Ҫ�R��ԇ��ˎƷƷ�N������1171 ��Ʒ�N���أ�ռ�ȸ��_81.9%���аlһ����ˎ��Ͷ��ɱ��dz��ߣ��������������ˎ�о��ɱ��Ľ��׃r��ͨ����3000 �fԪ���ϣ�һЩ�tˎ���ҵĹ�����ߣ��J���Ї��аlһ����ˎƽ���ɱ���2 �|Ԫ���ҡ���ˣ��Ј�����һЩ�аl�ӽ�β����Ո�@���ȴ����a��ˎ�������^�阷�^��δ���A�ڣ������Ո���ܻ����ӳ��أ��������F�����A�ڎ����^��ؓ��Ӱ푣����ڛ]�Ќ��|�аl��������I�o���Ǿ�_����
�_չ�R��ԇ��������Ո���ص�ˎƷƷ�N���l����
����1��ʳˎ�O���ֽ����_ʼ�����Ƅӷ���ˎһ�����u�r
����һ�����u�r��һ�N�l�_���Ҷ��V����õķ���ˎ�ИI�lչ�˜ʣ��e������������һ�ݽ������Ч���u�r�ϸ�����������е�ˎƷ��䛣��������ί���ͬ���u�r���ʵ�ˎƷ�������I��Q���Ƥ��“Orange Book”�����R��ʹ�������Ч�ķ���ˎ���ԭ��ˎ�ṩ����������ȫ���tˎ�ИIһ�����u�r�ı��档“��Ƥ��”���e��FDA ���ʵ�ˎƷ����ָ�����^“��Ч�Ը���”�������Ƅ���Ч�ԡ�ͬ�ɷ֮������Ƅ����ί���Ч�ԡ��������öȡ������Ч�Ƅ��ȣ������ί���Ч�ԛQ���˷���ˎ�cԭ��ˎ֮�g���Ƿ�һ�¡�
�������w�Ͽ�����������ƏVһ�����u�r�����@����̭���ϸ�ˎƷ��20 ���o70 ��������M���������Ч���u�r����̭�˼s6000 �N���ϸ�ˎƷ��1975 �꣬Ӣ���������u�r��1968��ˎƷ�����_֮ǰ���е�3 �f��NˎƷ��1998 ���ձ�������“ˎƷƷ�|���u�r����”��Ŀǰ���ڳ��m�M���У�����ԓ���̹������730 ����Ʒ�N��7000 �����aƷ���|��һ���Ժ���Ч�����u�r��400 �������_�˵�������̖��ע�N���aƷ�˳��Ј���
����2���������ƱO�ܙC��������齨�O
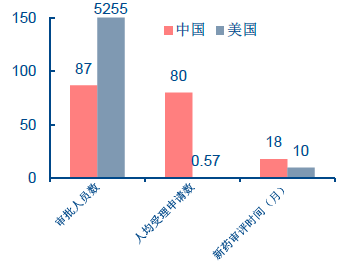
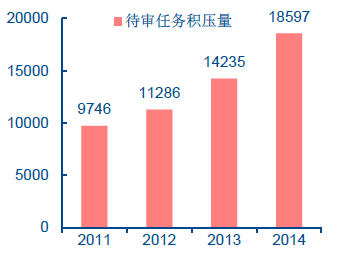
����һֱ�ԁ����҇�ʳˎ�O���������������㣬���g���u�˲Ň���ȱ��������FDA �Ќ��u�ˆT5255 �ˣ�ռFDA ���˔���45.6����ÿ�ꌏ����Ո��3000 �������҇�����ˎƷ���u���Ĺ���120 �ˣ����Ќ��I���u�T87 �ˣ�ÿ��ƽ������6000 �����u��Ո��ƽ��ÿ��ÿ��Ҫ��ɽ�80 ���Ŀ�Č��u�����H�ϸ����o����ɣ���ɴ�����Ո�e����2014 ���҇������΄շe����18597 ������2013 ������΄տ���������4362 �����ϝq��1/3���҇���ˎ�R��ԇ���Ո��������Ո�ķ����r�քe��90 ���150 �죬����ˎ��Ո�ķ����r�ޞ�160 �죬�����H��ˎ�R��ԇ���Ո��������Ո�ĵȴ��r�g�քe��8.1 ���º�1 ��룬����ˎ��Ո�ȴ��r�g�����L�_3 �ꡣ��ˣ��ӏ�ʳˎ�O���ֵ��˲Ž��O�������ˆT���Ɣ����������ˮƽ�ȿ̲��ݾ���
����ˎƷ���u�C����������
�Ї�ʳˎ�O���֔D����Ո��r����
����2016 ��3 ��19 �գ��ڇ���Ժ�lչ�о��������k���Ї��lչ�ߌ�Փ���ϣ�����ʳƷˎƷ�O����������ˎƷ���yƷע�Թ���˾˾�L�����S��ʾ��CDFA Ŀǰ�����M�ЙC�����ˆT�{�������P�ʂ乤�����e�nj�����ˎƷ���u���ģ�����ȫ����ļ���g�ˆT�������O�ܙC������������ͬ�r�����ط�ʳˎ�O��Ҳ�кܶ��˲Ž��O�ķe�O̽�������磬����ʡʳˎ�O�������˲���齨�O����2016 ��“����Ŀ��”֮һ��ȫ�����M�˲ŏ��֑��ԣ���ȫʡϵ�y�ȷӷ��Ҏģ�_չ�I����Ӗ��
����3�������ř࣬��߱O��Ч��
�������w�Ͽ����O�ܙC�������ĸĸ﷽����“�����з�”��������ǰ���ᵽ�Ą���ˎ�R�������˲顢����ˎһ�����u�r���ذ������ɇ���ʳˎ�O����ȫ��ؓ؟��ͬ�rͨ�^�����řಢ��һЩ�O�ܙ����·ŵ�ʡ����λ���@�������ڰl�]ʡ����λ���������w��߱O��Ч�ʡ�
2013 ���ԁ�����漰ˎƷ�ĸ����������
�����l�����T�ļ����Q���ă���2013 ��2 ������ʳƷˎƷ�O���������P���ˎƷ���u�����ĸ��Mһ���Ą�ˎ��µ���Ҋ�Mһ���ӿ섓��ˎ��u�����в��ַ���ˎ���Ȍ��u���ӏ�ˎ���R��ԇ��|���������Ą����ƃ�ͯ��ˎ���ƶ����״�ʩ��ע�f�{���2013 ��6 ������ʳƷˎƷ�O���������{�������t����е׃���������|�������wϵ�z��؟�����־��ȵ�����t����еע���C���͵�����w���\��ԇ����ӛ��׃��������헵ȹ����{����ʡ��ʳƷˎƷ�O���������T��ʩ2013 ��10 ������ʳƷˎƷ�O���������ӏ�ʡ���t����е���g���u�������O��ָ����Ҋ����ͨ�^2��3 ��ĕr�g��ʡ���t����е���g���u��������ߣ����u�ˆT���ú�����Ҏ���ƶȲ������ƣ���Ϣ��ˮƽ������ߣ��Ԍ��F�ƶȻ���Ҏ�������ƌW���Č��u����Ŀ��2013 ��12 ������ʳƷˎƷ�O���������P��2013 ��ȷ���ˎ�|��һ�����u�r�����о��΄յ�֪ͨ��ʡ��ˎƷ�O���������TҪ�ӏ��������V���ӆT���������P�����e�O���c��ԓ헹�����2014 ��2 ������ʳƷˎƷ�O�������������t����е�e��������ԇ�У��ڴ_�����ЮaƷ��ȫ����Ч��ǰ���£�ᘌ������t����е�O�õČ���ͨ��2014 ��4 ������ʳƷˎƷ�O���������P�����ò�������ˎƷ���������Ŀ�·����P������֪ͨ���^�������l��I��ʡ���t���C���N������ˎƷ�͵�һ���ˎƷ�����Č�ʩ�C�P�ɇ���ʳƷˎƷ�O�ܲ��T�·���ʡ��ʳƷˎƷ�O�ܲ��T2014 ��9 ������ʳƷˎƷ�O��������������һ���^���؏�ˎƷƷ�NĿ��������Ͷ�Y�������P��I��ע���u���аl�L�U�������M��Ͷ�Y���I�Q��2015 ��1 ������ʳƷˎƷ�O���������P������ȫ�挍ʩˎƷ��ӱO�ܹ�����֪ͨ�ӏ�ˎƷ���a���I��I��W�ĽM���������ӏ�ˎƷ��ӱO�ܹ������ȣ��M�쌍�F��ӱO��ȫƷ�Nȫ朗l���w2015 ��6 ������ʳƷˎƷ�O���������P�ڌ�ȡ�����·�����������헼ӏ������º�O�ܵ���Ҋ���_ȡ�����·ŵ�����������헣��yһҎ�������˜ʣ������н��������O���������䰸���º��u������O����Ϣ���_���������o���C��؟��2015 ��7 ������ʳƷˎƷ�O���������P����������ˎƷ���c���a�����ͱO�ܹ�����֪ͨ������ˎƷ����һ���ˎƷ�͵ڶ����ˎƷԭ��ˎ���c���a��������һ��е�ˎƷ����ƶ����WƷ���a�S�Ɍ����Č�ʩ�C�P��ʳƷˎƷ�O�ܿ����·���ʡ��ʳƷˎƷ�O���������T2015 ��8 ������ʳƷˎƷ�O���������P��ӡ�l�t����е���I��I��ּ��O������Ҏ����֪ͨʳƷˎƷ�O���������T�����t����е���L�U�̶ȡ��t����е���I��I�I�B���|������ˮƽ�����ط�Ҏ�������¼����aƷͶ�V��r�����أ����t����е���I��I�֞鲻ͬ��e�������ՌٵرO�ܵ�ԭ�t����ʩ�ּ��ӑB����2015 ��8 ������Ժ�P�ڸĸ�ˎƷ�t����е���u�����ƶȵ���Ҋ���ˎƷ�����˜ʣ����M����ˎ�|��һ�����u�r���ӿ섓��ˎ�Č��u�������_չˎƷ�����S�ɳ������ƶ�ԇ�c�����r�l��ˎƷ����������Ϣ�����MˎƷ�R��ԇ����2015 ��11 ������ʳƷˎƷ�O���������P��ˎƷע�Ԍ��u�����������ߵĹ�����߷���ˎ�����˜ʣ�Ҏ����������ˎ�Č��u�����������R��ԇ���Ո�Č��u����������ͬƷ�N���Ќ��u�����S��Ո�����ӳ��ز����ϗl����ˎƷע����Ո��2015 ��11 ������ʳƷˎƷ�O���������P�ڬF�ЏĘIˎ��ʹ�ù��톖�}��֪ͨ��ʩ�ĘIˎ���^�����ߣ����L�߂�l���ďĘIˎ���Y����Ч�ڣ��_�J�߂�l���ďĘIˎ�����^���ڏĘIˎ����ʹ�ú���2016 ��2 ������ʳƷˎƷ�O���������P�ڽ�QˎƷע����Ո�e�����Ѓ��Ȍ��u��������Ҋ��ӏ�ˎƷע�Թ������ӿ�����R���rֵ����ˎ���R���������ˎ���аl���У���QˎƷע����Ո�e����ì�ܣ�������Ȍ��u�����ķ������������Ҫ��2016 ��3 ������Ժ�P���_չ����ˎ�|���ͯ�Чһ�����u�r����Ҋ���_�u�r����͕r�ޣ��_�������Ƅ����xԭ�t�������x���u�r�������䌍��I���w؟�Σ��ӏ���һ�����u�r�����Ĺ������Ą���I�_չһ�����u�r���������������İ���wԭ�������У��D�d���H�����������Ϣ֮Ŀ�ģ������֙��О飬Ո��һ�r�gϵ�҂��Ļ�h�����]�䣺cidr@chinaidr.com�� -
- ���c�YӍ
- 24С�r
- ������
- ������
- �����Ϳ�x�ܷ��z���ۣ����̘I����������y�Ļ��ĬF�������_
- ��e؛�\ä�^�L�U��G7�������ֻ��������ܰ�ȫ�ډ�
- һ�l���a�I���ȡ���һ�����������ȡ� �ɶ��������l����ɫ�Lò��·�۳Ǵ����ӛ
- ��늡���ƽ�����У��W�m������ȫ�����������η���
- ��ܛ���Г��Ŀ���x��2025��ȇ��ҙn���ֿƼ��Ŀ�M����Ŀ���Ρ�
- �����ɣ����̓r���Ǹ����������ͳɱ����������o�Ǻ�
- ������Ʒʡ�X�������Sģ�ͣ��������ؘ���^���������B
- �a���ں��Ӱ壡�ӱ���W�y�����Ƽ������������`�����c��������
- ؐ�J�ѹ�Ӣ���ؽM�W�������h���L���I��ϵ�y������ȫ�o�n
- ��������Ƥ���������£�㼑г�M�@�����죡


��� | �Wվ��B | �P���҂� | �aƷ�c���� | �̘I���� | ����� | �������� | �̘I�YӍ | ϵ�҂� | ����朽� | �Wվ�؈D
���������������б����д��·ʮһ̖11̖�̄�4�ӡ��]����100141
�����\�I���ģ������н������g�_�l�^�P�Ƕ�·10̖��ؕr���V��C��12��
ȫ�����M��ԃ�ᾀ��400-680-5790 (7*24С�r�������ԃ�Ԓ��18411010258 ���棺010-58850975
�I����ԃ������17810330644�������� ����]����cidr#chinaidr.com(��#�Q��@) ����QQ��330291710
Copyright © 2009-2020 chinaidr.com, All Rights Reserved���a�I�lչ�о��W ������� �Wվ�䰸����ICP��11011445̖-2
���������������б����д��·ʮһ̖11̖�̄�4�ӡ��]����100141
�����\�I���ģ������н������g�_�l�^�P�Ƕ�·10̖��ؕr���V��C��12��
ȫ�����M��ԃ�ᾀ��400-680-5790 (7*24С�r�������ԃ�Ԓ��18411010258 ���棺010-58850975
�I����ԃ������17810330644�������� ����]����cidr#chinaidr.com(��#�Q��@) ����QQ��330291710
Copyright © 2009-2020 chinaidr.com, All Rights Reserved���a�I�lչ�о��W ������� �Wվ�䰸����ICP��11011445̖-2